Case Report
2015
September
Volume : 3
Issue : 3
Complete androgen insensitivity syndrome: Case based review article
Babulreddy Hanmayyagari
Pdf Page Numbers :- 130-133
Babulreddy Hanmayyagari1,*
1Department of Endocrinology, Krishna Institute of Medical Sciences, Kondapur, Hyderabad -500084, Telangana, India
*Corresponding author: Dr. H. Babul Reddy, Department of Endocrinology, Krishna Institute of Medical Sciences, Kondapur, Hyderabad -500084, Telangana, India. Mobile: 9985661434; Email: babulreddy78@gmail.com
Received 4 May 2015; Revised 15 June 2015; Accepted 22 June 2015; Published 30 June 2015
Citation: Babulreddy H. Complete androgen insensitivity syndrome: Case based review article. J Med Sci Res. 2015; 3(3):130-133. DOI: http://dx.doi.org/10.17727/JMSR.2015/3-025
Copyright: © 2015 Babulreddy H, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Complete androgen insensitivity syndrome (CAIS) a form of 46 XY disorder of sex development (DSD), usually presents with an inguinal hernia in an infant or a child and with primary amenorrhea in adult phenotypic female. We hereby report a case who reared as a female presented to us with primary amenorrhea and on further evaluation, diagnosed to have CAIS, then we also discuss in detail about this rare entity.
Keywords: complete androgen insensitivity syndrome; primary amenorrhea; gonadectomy
Full Text
Introduction
Complete androgen insensitivity syndrome (CAIS), Otherwise called as testicular feminization syndrome [1] is a rare form of 46 XY DSD inherited as X-linked recessive condition, due to mutations in androgen receptor. Up to 30% of cases are sporadic [2]. The estimated prevalence of AIS is between 1 in 20000- 99000 genetic males [3]. The responsible gene is located in the proximal, long arm of the X chromosome [4].
An affected individual has 46XY karyotype with normal differentiation of testes in utero. But a defect in the gene coding for the androgen receptor (AR) results in complete insensitivity to circulating androgens, thereby resetting in phenotypic female development [5]. Psychosexual orientation is feminine. There is, however, no uterus and only a partially formed vagina, and pubic and axillary hair is scant or absent. The increased testosterone production is secondary to increase in serum luteinizing hormone (LH) concentrations, resulting from resistance to the feedback effect of androgens on LH secretion at the hypothalamic-pituitary level. Both the frequency and amplitude of pulses of LH secretion are increased [6]. Serum follicle-stimulating hormone (FSH) concentrations are usually normal. Here a case of CAIS is reported subsequently the same was discussed in detail about this entity.
Case report
20-year-old female (Figure 1) was referred from gynecologist to the Department of Endocrinology, KIMS Hospital, for the evaluation of primary amenorrhea. She was born to the parents of non-consanguineous marriage. Full term normal delivery, 3rd in birth order, external genitalia were like normal female at birth with small inguinal swellings bilaterally. She had breast development at 12 years of age but history negative for any recurrent cyclical abdominal pain, acne, hirsutism, any visual field defects and for galactorrhea. No family member had similar complaints. On examination her weight was 42 kg, height was 164 cm, with normal upper to lower segment ratio of 0.9. There was no goitre, her sexual maturity rating (SMR) was tanner breast [3], pubic hair [3], axillary hair [1] (Figures 2, 3, 4). Local examination was normal with blind vagina, about 3x2cm palpable inguinal masses felt bilaterally (Figures 4, 5). Systemic examination including heart and lungs was normal.
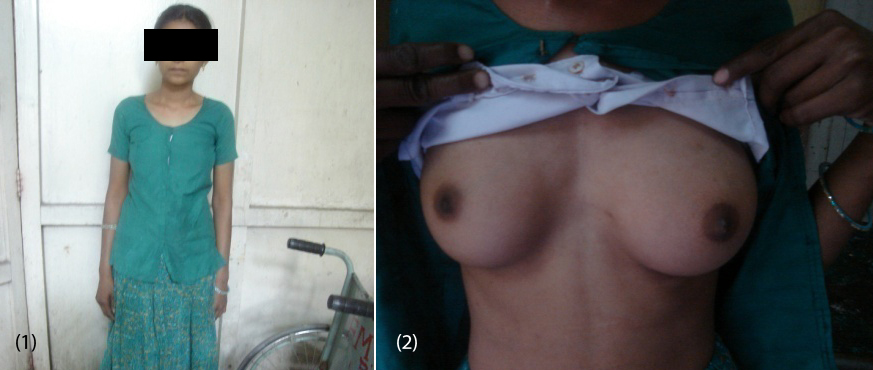
Figures: (1) Showing the patient. (2) Tanner breast stage 3.
Investigations
Routine biochemistry was normal. Serum LH was 37.26 miU/ml (1.5-12), and FSH was 7.14 miU/ml (1.6-10), testosterone 758.14ng/dl (female 0.2-0.6, male 3-6.5), estradiol 57.07 pg/ml (female 10-50), On ultrasound abdomen no mullarian structures were seen, bilateral gonads were seen in inguinal area with right gonad measuring 2.9 cm X 1.7cm, left gonad 2.5 cm X 1.5cm (Figure 6) and her karyotyping was 46 XY.
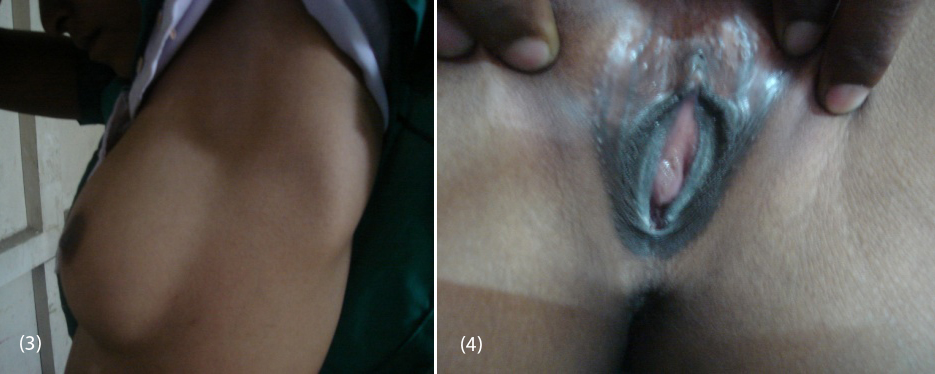
Figures: (3) Showing no axillary hair (Tanners stage 1). (4) Showing blind vagina.
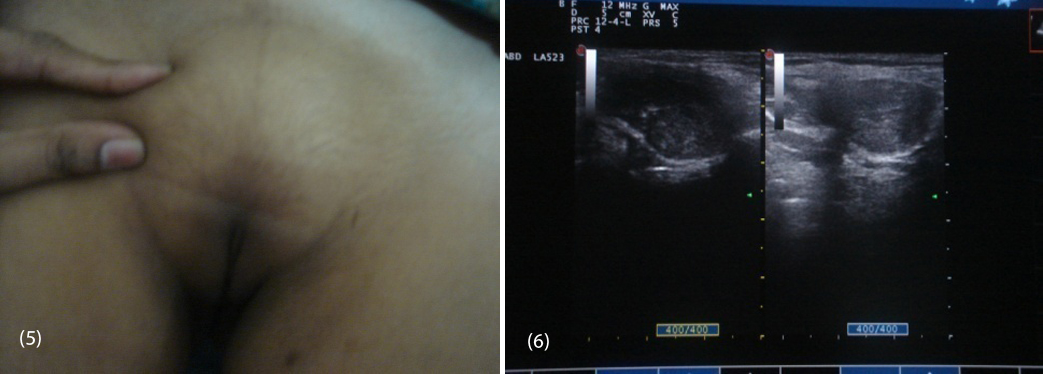
Figures: (5) Showing sparse pubic hair with palpable gonads. (6) USG showing bilateral gonads in inguinal area.
So patient was diagnosed to have 46 XY DSD with complete androgen insensitivity syndrome. She was counseled about the issues with this disorder and about the prospects of fertility, later she underwent bilateral orchidectomy (Figure 7a,b) and vaginoplasty. Post operatively the patient was put on estrogen replacement therapy and she was under regular follow up.
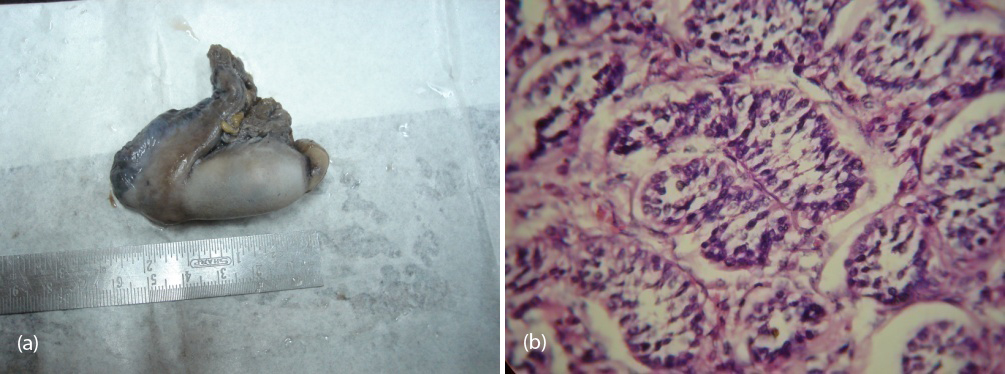
Figure 7a,b: Showing gross and histological confirmation of testis with seminiferous tubules.
Discussion
The typical presentation of CAIS in an infant or child is with inguinal hernia in a phenotypic female. In later period the most common presentation is that of primary amenorrhea with normal breast development. Examination reveals tanner breast development consistent with age with scant to absent axillary and pubic hair. Height is typically in the male mid parental height range and the genitourinary examination reveals normal external genitalia with rudimentary, blind ending vagina. Various locations of testes have been mentioned in patients with CAIS. One study out of 52 patients in whom complete androgen insensitivity syndrome diagnosed, 35 (67%) had abdominal, 16 had inguinal and one had labial testes [7]. The main differential diagnosis for CAIS are complete gonadal dysgenesis (Swyer syndrome) which is distinguished by poor breast development, short stature, 5α-Reductase deficiency can be distinguished by progressive virilisation after puberty & True hermaphrodism which can be differentiated by some degree of masculanization, presence of ovarian tissue on histology specimen.
The major concern with regard to cryptorchid testes (more often in abdominal than in inguinal testes) is the development of a neoplasia, usually a gonadoblastoma or a malignant dysgerminoma. The incidence of neoplastic formation is 52%, half of which are malignant, and usually occurs after puberty. The risk of developing malignancy increases with age, reaching 33% at the age of 50 [8, 9]. So, prophylactic gonadectomy is advised.
Even though much controversy exists about the timing of gonadectomy, most agree that subjects of CAIS have a normal pubertal growth spurt and feminize at the time of expected puberty and because the tumors do not usually develop until after this time [9], gonadectomy in women with CAIS can be delayed until sexual maturation is complete [10, 11]. Carcinoma in situ of the testis has been described in prepubertal girls with CAIS, but its importance is uncertain [12]. In rare situations, the convenience of combining gonadectomy with hernia repair or the need to relieve symptoms of discomfort from labial or inguinal testes justifies removal of the testes before puberty. Management of CAIS needs multidisciplinary approach including gonadectomy, surgical correction and detailed psychological counseling along with hormonal replacement. Limitations in our case are we have neither carried out a genetic analysis of androgen receptor in our index case nor screened her family.
Conclusion
Complete androgen insensitivity syndrome is a rare form of disorder of sex development. One should include this in the differential diagnosis of primary amenorrhea in a phenotypic female.
Conflict of interest
The authors declare no conflict of interest.
References
1. Morris JM. The syndrome of testicular feminization in male pseudo herma phrodites. Am J Obstet Gynaecol 1953; 65(6):1192–1211.
2. Hughes I, Deeb A. Androgen resistance. Best Pract Res Clin Endocrinol Metab 2006; 20(4):577–598.
3. Boehmer AL, Brinkmann O, Brüggenwirth H, van Assendelft C, Otten BJ, et al. Genotype versus phenotype in families with androgen insensitivity syndrome. J Clin Endocrinol Metab. 2001; 86(9):4151–4160.
4. Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, et al. Cloning of human androgen receptor and complementary DNA and localization to the X chromosome. Science. 1988; 240(4850):327–330.
5. Weiner JS, Teague JL, Roth DR, Gonzales ET Jr, Lamb DJ. Molecular biology and function of the androgen receptor in genital development. J Urol. 1997; 157(4):1377–1386.
6. Boyar RM, Moore RJ, Rosner W, Aiman J, Chipman J, et al. Studies of gonadotropin-gonadal dynamics in patients with androgen insensitivity. J Clin Endocrinol Metab. 1978; 47:1116.
7. Barthold JS, Rivers KK, Upadhyay J, Shekarriz B, McGinley JI. Testicular position in the androgen insensitivity syndrome: implications for the role of androgens in testicular descent. J Urol. 2000; 164(2):497–501.
8. Speroff L, Glass R, Kase N. Normal and abnormal sexual development in clinical gynaecologic endocrinology and infertility, Williams & Wilkins, Philadelphia, Pa, USA, 5th edition, 1994.
9. Hurt WG, Bodurtha JN, McCall JB, Ali MM. Seminoma in pubertal patient with androgen insensitivity syndrome. Am J Obstet Gynecol. 1989; 161(3):530–531.
10. Purves JT, Miles-Thomas J, Migeon C, Gearhart JP. Complete androgen insensitivity: the role of the surgeon. J Urol. 2008; 180 (4 Suppl):1716–1719.
11. Cheikhelard A, Morel Y, Thibaud E, Lortat-Jacob S, Jaubert F, et al. Long-term followup and comparison between genotype and phenotype in 29 cases of complete androgen insensitivity syndrome. J Urol. 2008; 180(4):1496–1501.
12. Cassio A, Cacciari E, D'Errico A, Balsamo A, Grigioni FW, et al. Incidence of intratubular germ cell neoplasia in androgen insensitivity syndrome. Acta Endocrinol (Copenh). 1990; 123(4):416–422.