Short Communications
2014
December
Volume : 2
Issue : 4
Haemophilic arthropathy
Sarath Chandra Mouli V, Pradeep H
Pdf Page Numbers :- 225-227
Dr. V. Sarath Chandra Mouli1,* and H. Pradeep1
1Department of Rheumatology, KIMS Hospitals, Secunderabad, Telangana, India
*Corresponding author: Dr. Sarath Chandra Mouli Veeravalli, MD (NIMS), MRCP (UK), Chief Rheumatologist, Department of Rheumatology, KIMS Hospitals, Secunderabad, Telangana, India. Email: chandramouli776@gmail.com
Received 14 July 2014; Revised 24 August 2014; Accepted 03 September 2014
Citation: Sarath Chandra Mouli V, Pradeep H. Haemophilic arthropathy. J Med Sci Res 2014; 2(4):225-227. DOI: http://dx.doi.org/10.17727/JMSR.2014/2-039
Copyright: © 2014 Sarath Chandra Mouli V, et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Acquired haemophilia (AH) is extremely rare and potentially fatal bleeding disorder and it is associated with significant morbidity and mortality rate between 7.9% to 22%. Upto 90% of people with AH experience bleeding. It is mainly due to the spontaneous development of auto antibodies that inhibit the coagulant activity of factor VIII (FVIII). Autoimmune disorders (16.6%) like Rheumatoid arthritis, Systemic lupus erythematosus (SLE), Multiple Sclerosis etc., may lead to trigger acquired inhibitors to Factor VIII. In the case report we described a 70-year-old male patient who has history of migratory joint pains and swellings for five months and on physical examination there was an echymotic rash over the left hand elbow and left groin extending into the left thigh and we reemphasize that even though a series of effective treatment options available, a through workup, early diagnosis and early treatment are important.
Keywords: Haemophilic arthropathy; Acquired Haemophilia; Rheumatoid arthritis
Full Text
Case presentation
A 70-years-old male from West Godavari district presented to Rheumatology OPD with a history of migratory joint pains and swellings for five months. Recently he developed pain, swelling and redness of left thigh, left knee and left ankle joints (Figure 1) for one week, with no history of trauma or fever. He was a cigarette smoker and chronic alcoholic but stopped two years ago. On physical examination there was an echymotic rash over the left hand elbow and left groin extending into the left thigh. His left knee was swollen but there was no tenderness or erythema.
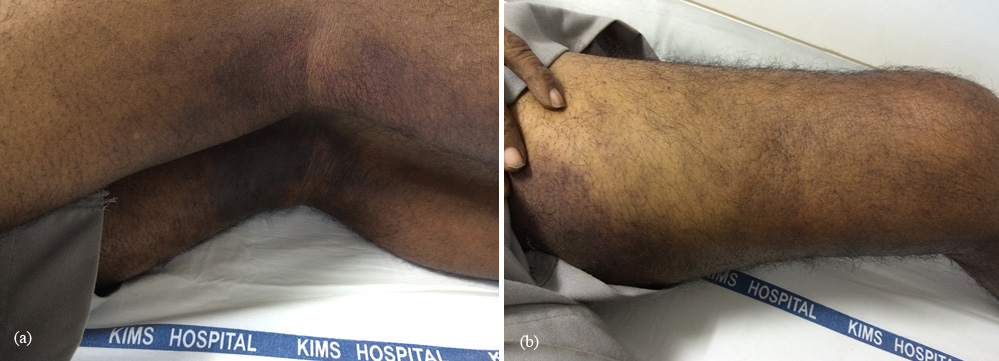
Figure 1a,b: The right leg with swelling and redness.
On laboratory investigations complete blood picture showed 5.3 gm% haemoglobin, 14500 cells/cumm; WBC, 3.68 lakhs/cumm platelets. Prothrombin test (PT/INR) was 13.3/1.12 sec, serum uric acid was 3.7 mg/dl and his activated partial thromboplastin time (coagulation method) was 132 sec, inhibitor screening (incubation method) was positive, factor VIII (FVIII) -1.0%, inhibitor to factor VIII 179.2 BU (Bethesda units). Complete urine examination was normal. High resolution ultrasound of thigh and left knee joint performed at 5 – 17 MHz linear transducer showed evidence of a large heterogeneous collection measuring 9.5 x 7.3 cm in the intramuscular planes of the medial aspect of the left upper thigh (haematoma) and synovial thickening with collection involving the left knee joint.
Acquired haemophilia
Acquired Haemophilia is extremely rare and potentially fatal bleeding disorder and it is associated with significant morbidity and mortality rate between 7.9% to 22%. Upto 90% of people with AH experience bleeding [1]. It is mainly due to the spontaneous development of auto antibodies that inhibit the coagulant activity of factor VIII (FVIII) [2, 3]. Such acquired inhibitors are clearly distinct from FVIII alloantibodies occurring in patients with congenital haemophilia. The development of autoantibodies against factor VIII leads to deficiency of factor VIII, which results in insufficient generation of thrombin by factor IXa and the factor VIIIa complex through the intrinsic pathway of the coagulation cascade.
There are certain circumstances that may lead to trigger acquired inhibitors to factor VIII which include postpartum period (2%), malignancies (14.6%), autoimmune disorders (16.6%) (Rheumatoid arthritis, SLE= Multiple Sclerosis, Autoimmune hypothyroidism, Sjogren syndrome, Myasthenia gravis etc.). It is reported that 40% of patients with acquired haemophilia are associated with above conditions. On other hand intake or overdose of anticoagulants agents, acquired vascular or platelet bleeding disorder or acquired deficiency of clotting factors and certain drugs like penicillin, sulphonamides, methyldopa, interferon alpha etc., [4 - 6] may rarely result in factor VIII autoantibodies. However, more than half of acquired haemophilia occurs without any underlying disease (idiopathic AH) [1, 7].
Acquired haemophilia has a global distribution and has a frequency rate of 1.3 to 1.5 cases/million persons/year. In India, it has been reported that the incidence of AH to be 0.8-1.52 cases/million persons/year [8]. Acquired haemophilia occurs in all racial groups and has no genetic inheritance pattern and is seen equally in men and women [9], although it is more prevalent in females with pregnancy and males constitute majority of patients over the age of 60 [10, 11].
Age at onset for AH is distributed in a biphasic patterns, with a small peak in young individuals, primarily in postpartum women and those with autoimmune diseases and a major peak in those aged 60-80 years of either sex. AH is uncommon in children younger than age 16, estimated at 0.045 cases/million persons/year. However, the prevalence rates are underrate due to its low frequency of incidence and it may be under diagnosis in elder people >85 years [1].
The clinical picture of AH is presented by severe bleeding which are spontaneous (mainly involving the skin (purpura) or mucosa) [6] they are usually painful and cause significant drop in haemoglobin level [12]. The detection of abnormal coagulation tests caused by circulating specific autoantibodies (inhibitors) is the characteristics of AH. In contrast with congenital haemophilia the bleeding patterns differ in AH. The distinctive feature of severe congenital haemophilia is hemarthrosis, this is unusual in AH. Prolonged activated partial thromboplastin time (APTT) is the most commonly reported laboratory finding [4] and patients are rarely diagnosed in the pre-symptomatic state through identifying prolonged APTT. Deaths are more frequent during the first few weeks after its appearance [4], median 19 days from diagnosis [13] and are mainly caused by gastrointestinal and/or lung bleeding, whereas intracranial and retroperitoneal bleeds predominate later [6].
Discussion and Conclusion
The case is reported a patient with acquired haemophilia with no family history of bleeding disorders and was critically ill. Due to its extreme severity, complexity of diagnosis, treatment and high mortality rate immediate consultation with experienced physician or haematologist should be done. Early diagnosis must be made to avoid delay in the treatment. We want to emphasize that even though a series of effective treatment options available, a thorough workup, early diagnosis and early treatment are important. Lack of familiarity with the disorder may lead to delayed diagnosis and sub optimal treatment. The lack of prompt and appropriate treatment in India often leads to major clinical complications, additional costs and even death.
Conflict of interest
The authors declare no conflict of interest.
References
1. Franchini MG, Gandini T, Paolantonio D, Mariani G. Acquired haemophilia A: A concise review. Am J Hematol. 2005; 80(1):55–63.
2. Shapiro SS, Hultin M. Acquired inhibitors to the blood coagulation factors. Semin Thromb Haemostas. 1975; 1:336–385.
3. Shulman NR, Hirschman RJ. Acquired haemophilia. Trans Assoc Am Physicians 1969; 82:338–397.
4. Green D, Lechner K. A Survey of 215 Non-hemophilic patients with inhibitors to factor VIII. Thromb Haemost. 1981; 45(3):200–203.
5. Morrison AE, Ludlam CA, Kessler C. Use of Porcine Factor VIII in the Treatment of Patients with Acquired Haemophilia. Blood 1993; 81(6):1513–1520.
6. Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, et al. Acquired haemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109:1870–1877.
7. Hay CR. Acquired Haemophilia. Baillieres Clin Haematol. 1998; 11(2):287–303.
8. Kashyap R, Choudhry VP, Mahapatra M, Chumber S, Saxena R, et al. Post partum acquired haemophilia: Clinical recognition and management. Haemophilia. 2001; 7(3):327–330.
9. Ma AD, Carrizosa D. Acquired factor VIII inhibitors: Pathophysiology and treatment”. Hematology: Am. Soc. Hematol. Educ. Program 2006; 432–437.
10. Cohen AJ, Kessler CM. Acquired inhibitors. Bailleres Clin Haematol. 1996; 9:331–354.
11. Bouvry P, Recloux P. Acquired haemophilia. Haematologica 1994; 79:550–556.
12. Windyga J, Chojnowski K, Klukowska A, et al. Polish recommendations on the management of acquired haemophilia. Med Prakt. 2011; 10:42–51.
13. Collins PW, Percy CL. Advances in the understanding of acquired haemophilia A: implications for clinical practice. Br J Haematol. 2010; 148(2):183–194