Case Report
2018
March
Volume : 6
Issue : 1
Peutz-Jeghers syndrome as an- emergency presentation
Sumana B, Rao VRK, Sahaja K, Swaroopa K, Kalyani B, Venkatesh B
Pdf Page Numbers :- 32-35
Sumana B1,*, Rao VRK1, Sahaja K1, Swaroopa K1, Kalyani B1 and Venkatesh B1
1Department of Radiology, Great Eastern Medical School and Hospital, Aditya Educational Society, Srikakulam Dist, Ragolu, Andhra Pradesh-532484, India
*Corresponding author: Sumana Bingi, Department of Radiology, Great Eastern Medical School and Hospital, Aditya Educational Society, Srikakulam Dist, Ragolu, Andhra Pradesh-532484, India. Email: sumana.roses7@gmail.com
Received 16 October 2017; Revised 11 December 2017; Accepted 20 December 2017; Published 29 December 2017
Citation: Sumana B, Rao VRK, Sahaja K, Swaroopa K, Kalyani B, Venkatesh B. Peutz-Jeghers syndrome as an- emergency presentation. J Med Sci Res. 2018; 6(1):32-35. DOI: http://dx.doi.org/10.17727/JMSR.2018/6-6
Copyright: © 2018 Sumana B et al. Published by KIMS Foundation and Research Center. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
An 18-year-old male patient presenting with mucocutaneous lesions of the face, chronic recurring abdominal pain for two years and recent swelling in the right hypochondrium for two days was investigated by CT scan. Multiple polypoidal lesions within the proximal jejunal loops were noted in addition to a large mass causing intussusception in the descending colon. Surgical decompression, imaging features and histological findings confirmed the diagnosis of Peutz-Jeghers syndrome.
Keywords: Peutz-Jeghers syndrome; hamartomatous polyps; mucocutaneous pigmentation; intussusception; malignancy; endoscopy
Full Text
Introduction
Peutz-Jeghers syndrome (PJS) is a rare polyposis syndrome with autosomal dominant inheritance having multiple non neoplastic hamartomatous polyps of varying sizes and shapes mostly in children in the age group of 11 to 13 years. The prevalence rate is from 1 in 1,00,000 to 2,80,000. Small intestine predominantly ileum, colon and stomach are involved in the descending order. 25 to 50 % of adolescents and childhood patients having polyposis in the small and large intestine have associated PJ syndrome. Only 12 cases were diagnosed as PJS polyps out of a total of 46 gastrointestinal polyps in a series of 44,615 surgical specimens during a period of five and half years. Pigmentation was lacking in less than 5% of patients with PJS and similarly, less than 5% with pigmentation lack polyps [1-3]. The incidence of intussusception in PJS is not clear in the literature. Characteristically the syndrome is associated with melanin pigmentation of mouth, fingers and toes. This article discusses the imaging, clinical and histological features of PJS with intussusception as an emergency presentation.
Case report
A male patient of 18-years age, presented with chief complaints of pain abdomen and swelling in the left hypochondrium since two days. Pain was of sudden onset, intermittent, colicky in nature, aggravated on straining and relieved with rest over the past two years occurring every 10 days. Physical examination revealed tenderness over the left hypochondrium with a well-defined firm oval swelling measuring 4 × 5 cm without any visible peristalsis. Occasionally the patient had blood stained mucoid stools. Physical examination revealed pallor, pigmented lesions over the face, around the lips, buccal mucosa and bilateral pedal oedema (Figure 1). Plain and contrast enhanced computed tomography demonstrated multiple intraluminal enhancing polypoidal lesions in the proximal jejunal loops. A large homogeneously enhancing mass measuring 83 × 43 mm was noted in the descending colon. Pericolic fat planes were clear. There was no retroperitoneal or mesenteric lymphadenopathy (Figure 2).
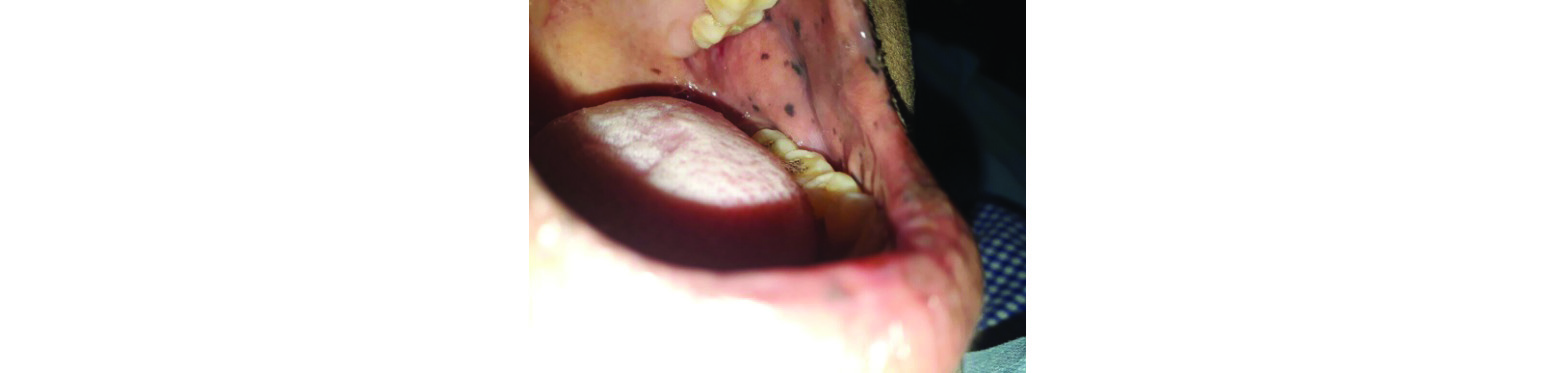
Figure 1: Clinical photograph - Pigmentation is seen in the buccal mucosa.
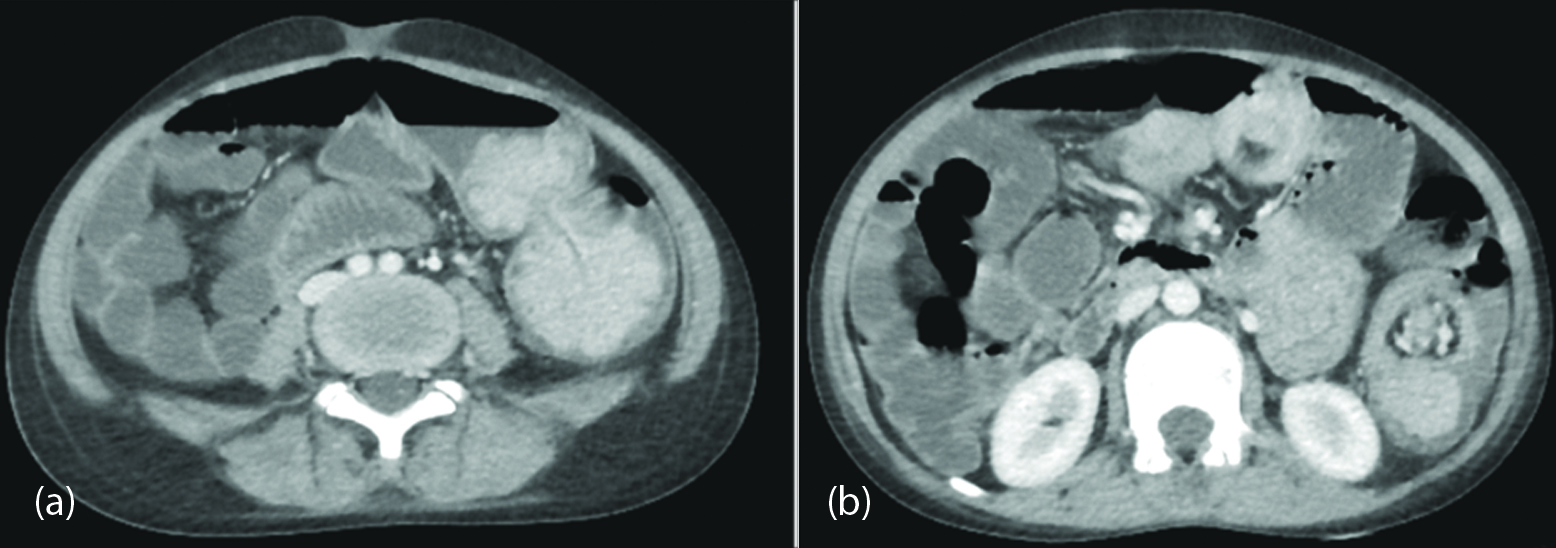
Figure 2: Contrast enhanced CT scan axial views, (a) Enhancing large polypoid intraluminal mass lesions in the jejunum are observed; (b) Telescoping jejunal loop is appreciated as intussusception.
Exploratory laparotomy revealed multiple polyps in the duodeno-jejunal junction, distal jejunum and ileum and a large polyp with lead point in the jejunum causing intussusception. Resection and end to end anastomosis was performed (Figure 3). Postoperative period was uneventful. Histopathology confirmed villoglandular polyp with low grade dysplasia having stratified nuclei and hyperchromatism without any invasive features (Figure 4).
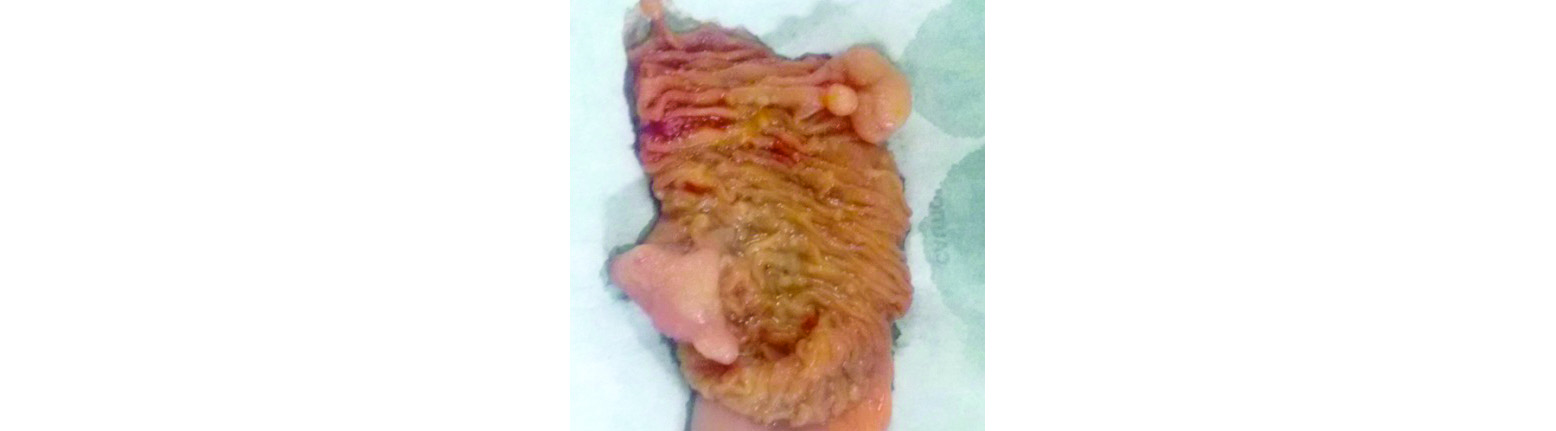
Figure 3: Specimen photograph - Multiple conglomerate polyps of varying sizes are noted.
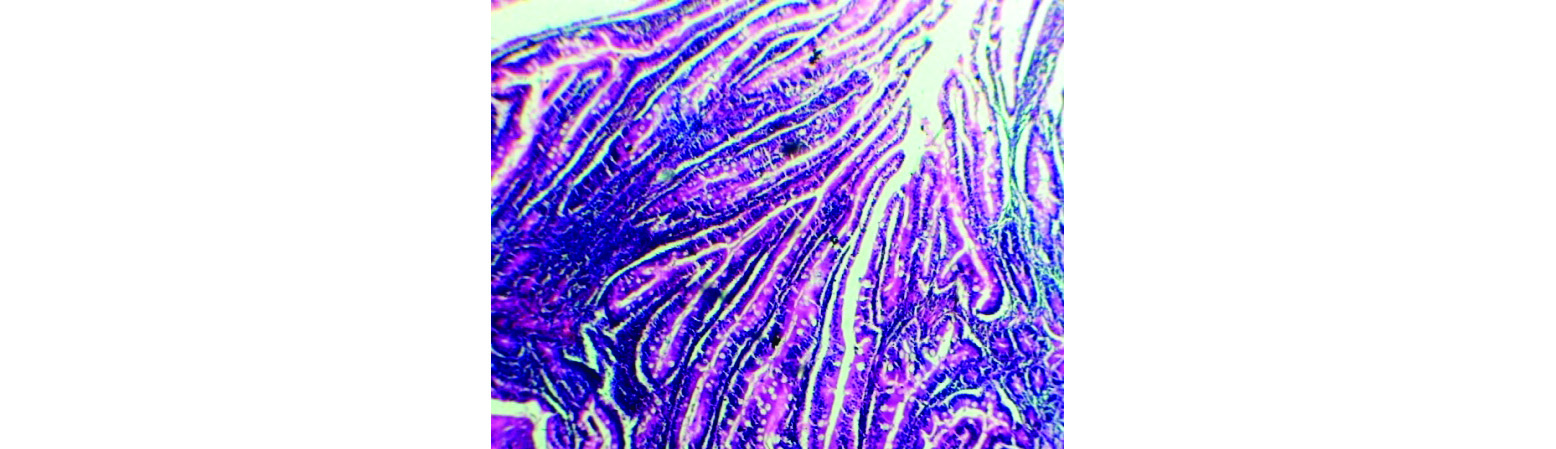
Figure 4: Photomicrograph - H & E 4X, a portion of polyp shows villi and glandular formation of mucosa.
Discussion
PJS is diagnosed in patients of second or third decade presenting with abdominal pain, rectal bleeding, intestinal polyps and obstruction due to intussusception. Three or more polyps with history of mucocutaneous pigmentation in any family member with polyposis are the diagnostic criteria laid down by the World Health Organization [4]. The gene responsible for PJS is located on chromosome 19p 13.3 [5]. PJS has two components: hamartomatous polyp involving the gastrointestinal tract and mucocutaneous pigmentation [6, 7]. The melanotic pigmented lesions do not turn malignant with a hamartomatous origin and generally fade during adolescence. The hamartomatous polyps occur with varying sizes from <1 cm to > 3.5 cm in diameter, pedunculated or sessile, commonly involving the small intestine predominantly ileum, colon and stomach in the descending order. They are known to occur in extra intestinal sites like kidneys, gall bladder, bronchial tree, ureters etc. These polyps may ulcerate and lead to chronic anaemia. PJS is associated with increased risk of intussusception, adenocarcinoma and other extra intestinal malignancies in the breast, pancreas, ovary, testis, lung and uterus. Histologically, continuity of muscularis mucosa in a branching pattern helps in distinguishing from other polyposis such as Cronkite Canada, juvenile polyposis and Cowden’s disease. Pseudo invasion due to epithelial misplacement possibly leads to recurrent bowel obstruction or intussusception. Imaging features of PJS are typical on barium studies, USG, CT and MRI. Intra operative enteroscopy (IOE) and double balloon enteroscopy (DBE) combined with video capsule enteroscopy (VCE) are gold standards for investigation. However, rapid capsule transit, poor luminal distension and restricted tumor size evaluation are well known limitations of VCE [8]. A longer symptom free interval is provided by complete polypectomy on DBE or IOE [9-11].
Conclusion
Mucocutaneous lesions associated with multiple enhancing polypoid intraluminal lesions observed on CT scan or endoscopy are diagnostic features of PJS. Intussusception in adult patients with pigmented mucocutaneous lesions should be suspected of PJS. Capsule endoscopy or MR enteroclysis, if available are performed for follow up. Screening of patients and first degree relatives is necessary for surveillance of this rare entity to avoid an increased risk of mechanical obstruction or malignant transformation.
Acknowledgment
The departments of Surgery and Pathology for their valuable contribution to the diagnosis and management of the patient.
Conflicts of interest
There are no conflicts of interest.
References
[1] Akshay BK, Prema M, Suvradeep M, Babu RT, KL Narasimha Rao. Solitary Peutz–Jeghers Polyp of Jejunum: A rare cause of childhood intussusception. J Indian Assoc Pediatr Surg. 2017; 22(4): 245–247.
[2] Tamanna C, Suraiya E, Ferdousy B, Md. Taufiq, Mohammed K. Peutz-Jeghers polyp: A retrospective study on twelve cases received at the Department of Pathology, Bangabandhu Sheikh Mujib Medical University. BSMMU J. 2012; 5(1):12–17.
[3] R Prathipaa, J Thanka, M Susruthan, Lawrence D Cruze. A Case of Peutz-Jeghers Syndrome with multiple intussusceptions. Annals of Pathology and Laboratory Medicine. 2016; 3(6):288–291.
[4] Girvin S, Glancy M, Dunlop M. Peutz-Jeghers syndrome: A case report and discussion of surveillance recommendations. Eur J Radiol Extra. 2007; 62(3):81–84.
[5] Mehenni H, Blouin JL, Radhakrishna U, Bhardwaj SS, Bhardwaj K, et al. Peutz-Jeghers syndrome: Confirmation of linkage of chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet. 1997; 61(6):1327–1334.
[6] Živković V, Pejović S, Nagorni A, Petrović B, Petrović A et al. Heriditary hamartomatous gastrointestinal polyposis syndrome. Scientific Journal of the Faculty of Medicine in Nis 2010; 27(2):93-103.
[7] Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syndrome: Diagnostic and therapeutic approach. World J Gastroenterol. 2009; 15(43):5397–5408.
[8] Gupta A, Postgate AJ, Burling D, Ilangovan R, Marshall M, et al. A prospective study of MR enterography versus capsule endoscopy for the surveillance of adult patients with Peutz-Jeghers syndrome. AJR Am J Roentgenol. 2010; 195(1):108–116.
[9] Kopacova M, Bures J, Vykouril L, Hladik P, Simkovic D, et al. Intraoperative endoscopy: Ten years experience at a single tertiary center. Surg Endosc. 2007; 21(7):1111–1116.
[10] Lin BC, Lien JM, Chen RJ, Fang JF, Wong YC. Combined endoscopic and surgical gtreatment of Peutz- Jegehrs syndrome. Surg Endosc. 2000; 14(12):1185–1187.
[11] Kopacova M, Rejchrt S, Tacheci I, Bures J. Hyperamylaemia of uncertain significance origin associated with oral double balloon enteroscopy. Gastrointestinal Endosc. 2007; 66:1133–1138.